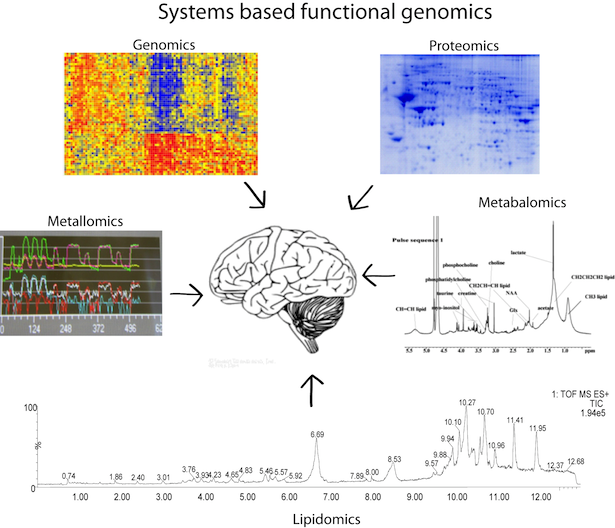
 During my PhD, I adopted a 'top-down' mult-omic systems biology approach to understand schizophrenia, a complex neuropsychiatric disease. I used genomics data from affymetrix-microarray chips, proteomics data from 2-D gel and mass spectrometry, lipidomics data from lipid mass spectrometry, metallomics data from laser ablation inductively coupled plasma mass spectrometry and metabalomics data from NMR to undetstand the disease mechanism and to identify its signatures. One of the main advantage of this multi-omic approach is that it gives a comprehensive and global picture of the disease mechanism. The challenge is to be able to integrate data from all these platforms and mine critical information. But there is no other significant methodology to understand a complex, non-mendelian disease like schizophrenia which have non defined phenotype and where genetic llinkage studies have associated it with a plethora of causative factors. The main results of this study indicated abnormality in energy metabolism and oxygen uptake pathways in schizophrenia. These results were novel findings and lead to new l approaches to understand the causative mechanisms of the disease. Pubmed. During my PhD, I adopted a 'top-down' mult-omic systems biology approach to understand schizophrenia, a complex neuropsychiatric disease. I used genomics data from affymetrix-microarray chips, proteomics data from 2-D gel and mass spectrometry, lipidomics data from lipid mass spectrometry, metallomics data from laser ablation inductively coupled plasma mass spectrometry and metabalomics data from NMR to undetstand the disease mechanism and to identify its signatures. One of the main advantage of this multi-omic approach is that it gives a comprehensive and global picture of the disease mechanism. The challenge is to be able to integrate data from all these platforms and mine critical information. But there is no other significant methodology to understand a complex, non-mendelian disease like schizophrenia which have non defined phenotype and where genetic llinkage studies have associated it with a plethora of causative factors. The main results of this study indicated abnormality in energy metabolism and oxygen uptake pathways in schizophrenia. These results were novel findings and lead to new l approaches to understand the causative mechanisms of the disease. Pubmed. |
One of the main disadvantages of the 'omic' systems biology approach is that it gives an overall 'static' picture of cellular mechanisms. Many disease mechanisms have been attributed to abnormalties in the dynamics of protein-protein interactions which can be understood only through quantitative experiments of protein-protein interactions and mathematical modelling. This approach is called 'bottom-up' systems biology. For part of my post doctoral work, I investigated the interaction of 3 proteins using quantitative mass-spectrometry and NMR and did mathematical modelling to predict the interactions. The system I looked at was a modifying, de-modifying cyclic protein phosphorylational and de-phosphorylational mechanism. These simple cyclic systems have been investigated in the past but we investigated modification/de-modifications on more than one site. In such systems, where in spite of all the molecular details of the interactions are known, are difficult to model and predict. The complexity of such systems arise from the multisite modifications. Proteins are generally percieved as 'on/off' switches when they are modified and de-modified in one site but when proteins undergo modificaitons on more than one site they can exsist in multiple intermediate states as well resulting in a distribution of protein states. It is this distribution of protein states that gives rise to complexity in protein-protein interaction dynamcis.
|
 |
|
The identification and quantitation of the phospho-forms of proteins is not a trivial task as the technology is not yet available. However, we found a way to arrive at the distribution of phospho-forms of proteins with a small number of phosphorylational sites, integrating results from protein-based MS, peptide-based MS with AQUA quantitation and NMR. Pubmed. |

| Using the above mentioned techniques, we have shown as a proof of principle that it is possible to calculate the the distribution of all the 16 phospho-forms of Erk, a key signaling protein with 4 phosphorylational sites (2 of which were novel findings identified by us). Hence in principle this methodoloy could be extended to all other proteins. | 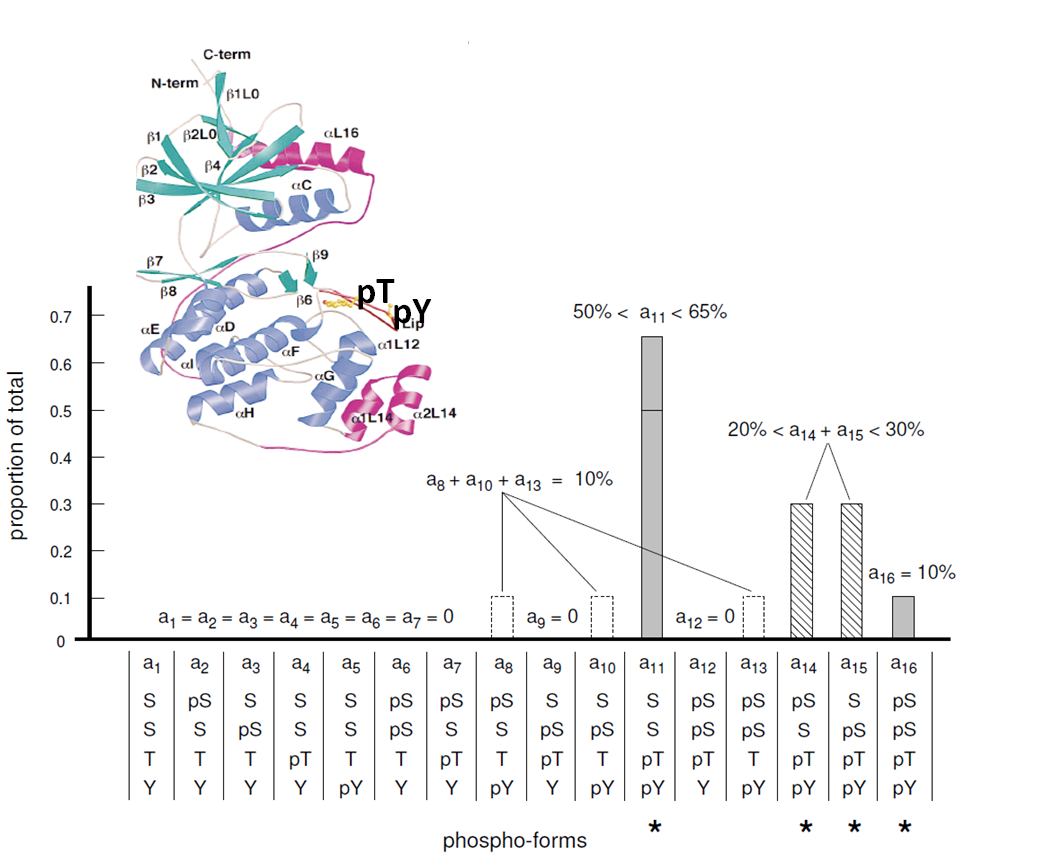 |
|